在最近发表于著名期刊《神经病理学学报》(Acta Neuropathologica)的研究中,瑞典卡罗林斯卡学院和意大利米兰大学的研究人员在人类神经元中发现了一种基因,可以抵御致命疾病肌萎缩侧索硬化(ALS)和脊髓性肌萎缩(SMA)中运动神经元的变性退化。在这些疾病的动物模型中,基因疗法被证明可以防止细胞死亡并延长寿命。
5 v5 J# S) J4 B: m6 F9 I8 `
/ g0 A0 q# Z1 o
ALS和SMA以运动神经元逐渐丢失为特征。由于这些神经元控制着身体所有的随意肌,其丢失会导致患者肌肉萎缩、无力和瘫痪麻痹。不过,有些运动神经元比其他的更敏感,例如脊髓中的神经元就对变性退化非常敏感,而脑干里负责移动眼睛的动眼神经元则拥有高度抵抗性。
M: O$ _! [, ]0 U0 Z) K! q! {( m8 M
研究人员发现的基因名为Synaptotagmin 13 (SYT13)。与脊髓中敏感的运动神经元相比,该基因在小鼠、大鼠和人类的动眼神经元中相对丰富。SYT13编码一种属于膜蛋白的蛋白质。
" m/ g; g0 E" X2 b# w+ \, h
保护作用
- j1 [/ a& V' E+ i: J
研究人员将取自ALS和SMA患者的诱导多能干细胞(iPS细胞)培育为人类运动神经元,然后针对这些运动神经元进行实验。研究人员发现,SYT13的引入能够通过减少内质网(ER)应激并阻断程序性细胞死亡来保护细胞免于变性退化。无论疾病的遗传病因如何,该基因都具有保护作用。
, ]0 J* J. J7 ^. O" j) `5 W
% y {) e% a' N# }# L3 P6 u2 j
* U9 ~; z* ]* M: I: w2 Q9 r: `
该研究资深作者之一、卡罗林斯卡学院神经科学系研究员Eva Hedlund说:“从治疗的角度来看这是非常有用的,因为90%的ALS患者神经丢失的机制很大程度上是未知的。”
7 Z c5 [& D" O4 c! P+ e# t2 e8 u7 @
在ALS和SMA动物模型中进行基因治疗实验后,研究团队发现,SYT13的引入可以避免这两种疾病中敏感的运动神经元变性退化。接受治疗的ALS和SMA小鼠,寿命分别延长14%和50%。
* q: Z. y) M; n, {4 S
有前景的候选基因疗法
' f% R( c6 d; y
该研究第一作者、米兰大学法拉利中心研究员Monica Nizzardo 说:“我们的结果提示,SYT13是治疗运动神经元病患者非常有前景的候选基因疗法。”
: W$ N+ @$ h$ F2 S P5 C. ~
目前,ALS缺乏有效的治疗方法,只有10%的家族遗传性患者病因较为明确。另一方面,SMA是由运动神经元生存1 (SMN1)基因突变导致的。两种靶向SMN1的新SMA疗法最近获得批准,结果很有希望,但疗效取决于治疗时间和疾病严重程度。
9 V8 }( M6 m1 I% g
" Q# J. u; n: z; @" s4 e$ A2 \3 _: }
图:Eva Hedlund实验室中从iPS细胞生成的人类运动神经元
! N O% v3 a# {7 ^! L; D- d
该研究另一位资深作者、米兰大学法拉利中心研究员Stefania Corti说:“对于SMA的新的补充疗法和能够帮助所有ALS患者的治疗形式,是目前迫切需要的。”
, X h E H0 m% M) S$ B2 k" L' b
寻找更多的治疗靶点
' r, K K u! s: ^
Eva Hedlund说:“我们将继续寻找抵抗性运动神经元特有的其他因素,从而识别更多的潜在治疗靶点。”
8 [+ f# F ?- {
- O6 g- E. t! ~! J4 c
& ]5 \5 U0 G! S5 I1 r% ?; C. Q





 发表于 2020-3-14 22:15:34
发表于 2020-3-14 22:15:34




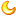
 发表于 2020-3-14 23:45:00
发表于 2020-3-14 23:45:00
{kind=link}